Characterization of Codon Usage Bias
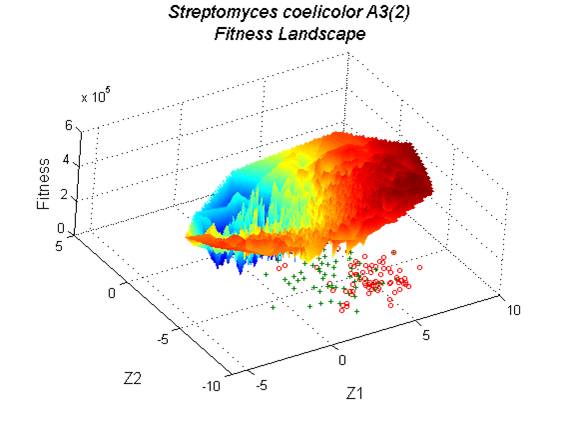
Genomic sequencing projects are an abundant source of information for biological studies ranging from the molecular to the ecological in scale; however, much of the information present may yet be hidden from casual analysis. One such information domain, trends in codon usage, can provide a wealth of information about an organism’s genes and their expression. Degeneracy in the genetic code allows more than one triplet codon to code for the same amino acid, and usage of these codons is often biased such that one or more of these synonymous codons is preferred. Detection of this bias is an important tool in the analysis of genomic data, particularly as a predictor of gene expressivity. Methods for identifying codon usage bias in genomic data that rely solely on genomic sequence data are susceptible to being confounded by the presence of several factors simultaneously influencing codon selection. We have developed novel techniques for removing the effects of one of the more common confounding factors, GC(AT)-content, and of visualizing the search-space for codon usage bias through the use of a solution landscape.